Leave No Development Candidate Behind
We make small molecule drug discovery faster, smarter, and more affordable. Deep Origin transforms complex simulations into intuitive workflows, bringing cutting-edge computational power to every scientist's fingertips.
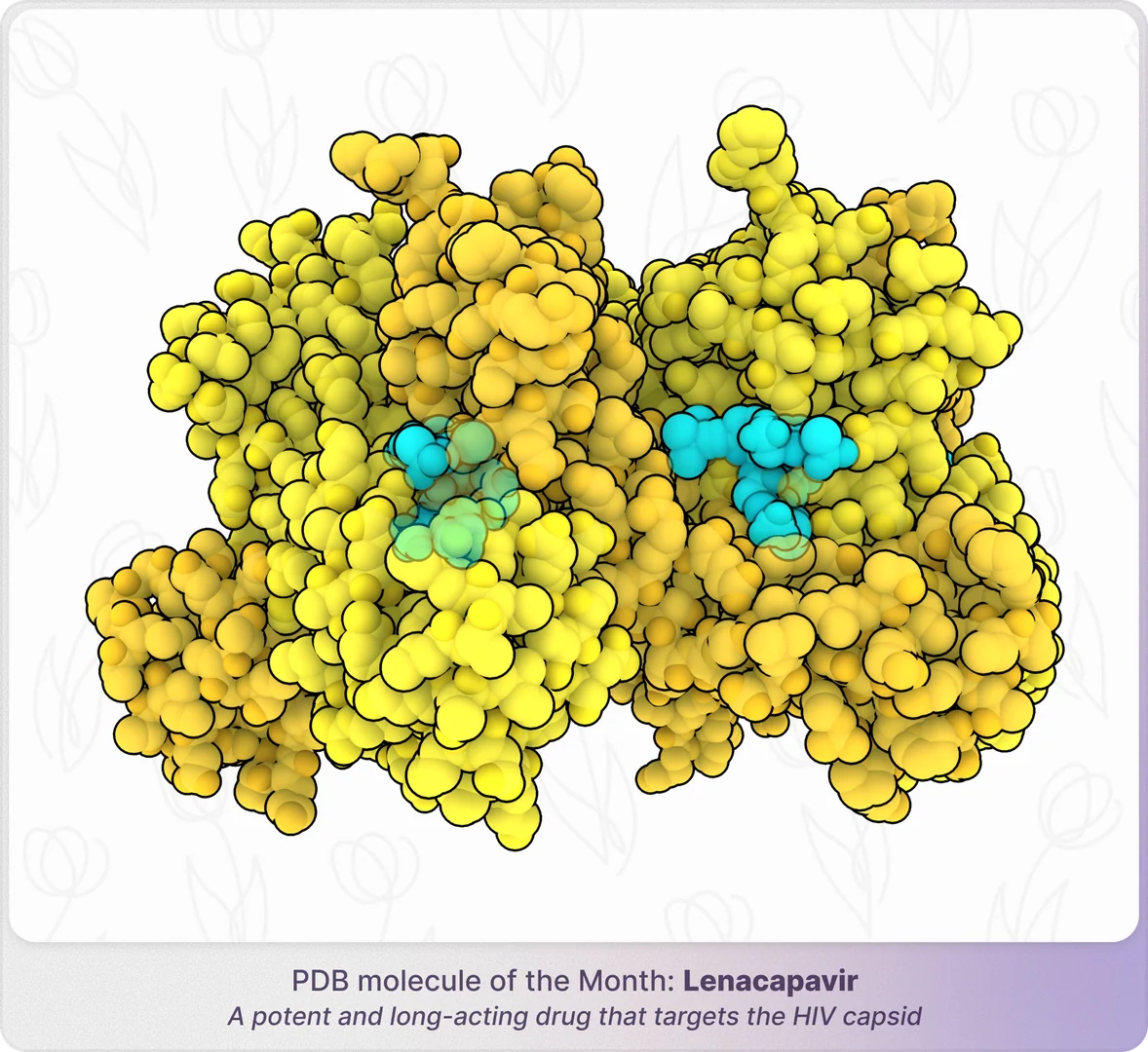
We combine molecular physics with AI-driven acceleration to revolutionize in silico drug discovery. Our intuitive AI assistant requires no computational expertise, making cutting-edge research accessible to all. Backed by a world-class team of computational scientists, medicinal chemists, physicists, and biologists, we tackle the toughest challenges in drug discovery.
Searching for a drug like a needle in a haystack? We'll find it for you.
Small Molecule Drug Discovery Tools Your Whole Team Can Use
Balto AI
No-code chemistry, docking, and drug discovery — made simple. Ask questions in plain English and get actionable results.
- Search PDB, ChEMBL, UniProt, PubChem, and patents
- Predict ADMET properties and drug-likeness
- Perform fast, high-accuracy ligand docking
- Interactive 2D/3D molecular visualization

# Find binding pockets
protein, pockets = pocket_finder_client.find_pockets(
protein, num_pockets=10, pocket_min_size=30)
# Dock ligands to protein
report = docking_client.dock(
protein=protein,
ligands=ligands,
pocket_data=pocket_data
)
# Get ADMET predictions
for result in report.results:
result.top_ligand.admet_properties()Deep Origin API
Code-first drug discovery — docking, free energy calculations, and molecular analysis at scale. Full programmatic control.
- Protein preparation and ligand analysis
- Binding pocket detection and docking
- ABFE/RBFE free energy simulations
- Process millions of molecules at scale
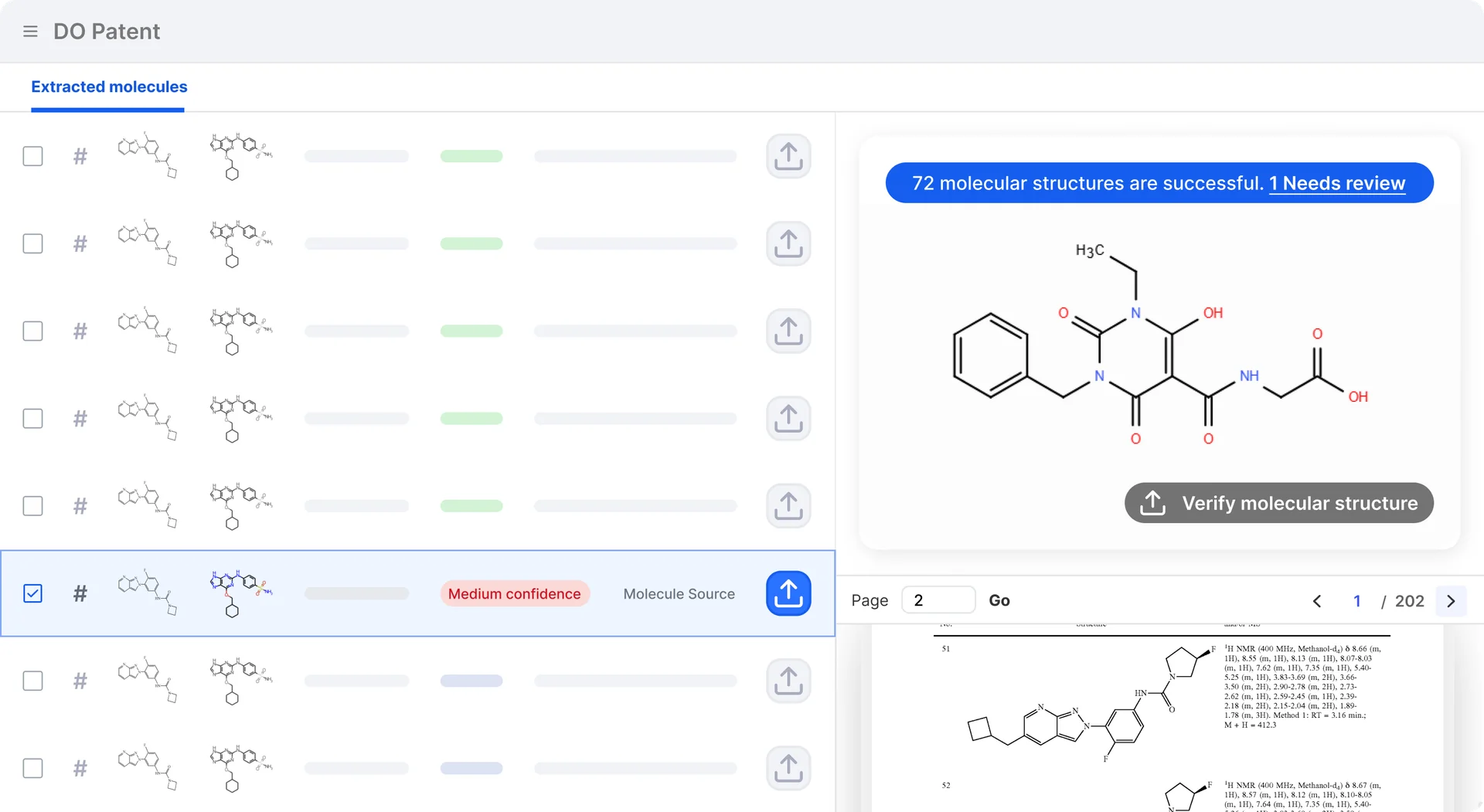
DO Patent
Extract molecular structures from patents and papers with 98%+ accuracy. Stop redrawing — start analyzing.
Why Deep Origin?
The platform built for modern drug discovery
Physics-Driven Accuracy
Our simulations are grounded in molecular physics, not just pattern matching. Get results you can trust.
Explore our scienceEnterprise Scale
Process millions of molecules. Our infrastructure scales with your ambitions.
Learn about partnershipsTools Your Whole Team Can Use
Our AI assistant guides you through complex workflows. Ask questions in plain English and get actionable results.
Try Balto for free100x Faster
What used to take weeks now takes hours. Accelerate your discovery timeline.
Take DO Patent for a spinSecure & Compliant
Your data stays yours. SOC-2 compliant infrastructure with end-to-end encryption.
Explore our science
Accelerate Your Small Molecule Drug Discovery Pipeline
From target identification to lead optimization, we combine physics-based accuracy with AI speed to transform how you discover drugs.
Latest Resources
Insights from our team on drug discovery, computational chemistry, and scientific innovation.
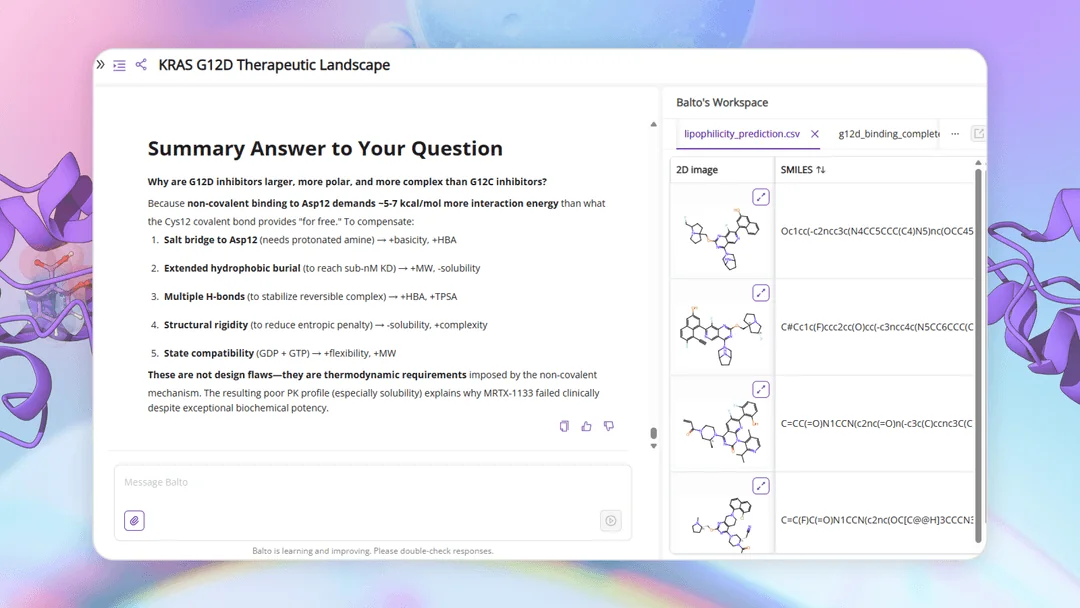
Finding Promising Molecules and Biological Background With Balto
Read articleWe Built a 98% Accurate Full Molecule Data Extractor for PDFs, Now You Can Use It
Ginkgo Bioworks partners on Deep Origin-led team to develop new tools for predicting drug safety
Out-of-the-Box GenAI for Medicinal Chemistry
A Blind Spot in Drug Discovery: Pocket Finding
Accelerating Drug Discovery with Physics-Informed Machine Learning
Practical and Accurate Free Energy Calculations using Neural Network Potentials
Connect with us
Get in touch to inquire about our solutions, discuss partnerships, join our team, or learn more.
We are excited to simulate life with you.