Drug discovery begins with an idea — a hunch that a certain pathway or protein could be the key to treating disease. But between that spark and a promising lead compound lies a swamp of friction: hours spent cross-searching literature, databases, and binding studies, just to confirm what’s already known.
Medicinal chemists routinely jump between a half-dozen disconnected tools: PubChem for physicochemical data, ChEMBL for assay results, PDB for binding evidence, Google Scholar for context. Each click leads to another tab, another export, another round of manual comparison.
That process is powerful — but feels pretty painful and slow for 2026.
I want to step you through what this process looks like with Balto. We’re going to find a promising molecule and its biological background, with thorough analysis at every step in less than an hour.
If you follow along through the walkthrough, you’ll see that Balto doesn’t replace scientific judgment. It lays out all the pertinent information, provides clarifications, and helps you chart the best path forward.
Ask a question, and Balto draws from curated sources like ChEMBL, PubChem, and literature databases, linking mechanistic data, binding affinities, and even known resistance mutations.
It’s as if every specialized search engine were already talking to each other, laying out information for and with you.
The result: scientists can move from “What if this target matters?” to “Here are the best candidate molecules to test” without ever leaving chat.
(You can then perform simulation, docking, and prediction of properties like ADMET directly in chat as well, but those are subjects for other walkthroughs.)
This first video below shows how that process works in practice. But let me warn you, Balto goes deep “into the weeds” (in a good way). Here we’re primarily exploring Balto’s agentic research capabilities. It can get a little long winded (and extremely thorough). So if you want to nerd out about the current cutting edge of AI-aided drug discovery iteration take a look at the Balto chat itself.
Anyways, onwards…
Let’s identify a promising small molecule with supporting biological evidence for a new drug target in less than an hour.
Step 1 — Exploring the biological landscape
Our initial prompt for Balto:
Summarize the current biological and therapeutic landscape around KRAS G12D as a drug target.
Include:
- Validated mechanisms of inhibition (e.g., covalent, allosteric, protein-protein disruption)
- Key signaling pathways involved
- Known clinical or preclinical failures and resistance mechanisms
- Open questions or emerging strategies that appear promising but underexplored
Please prioritize recent primary literature and reviews and cite your sources.
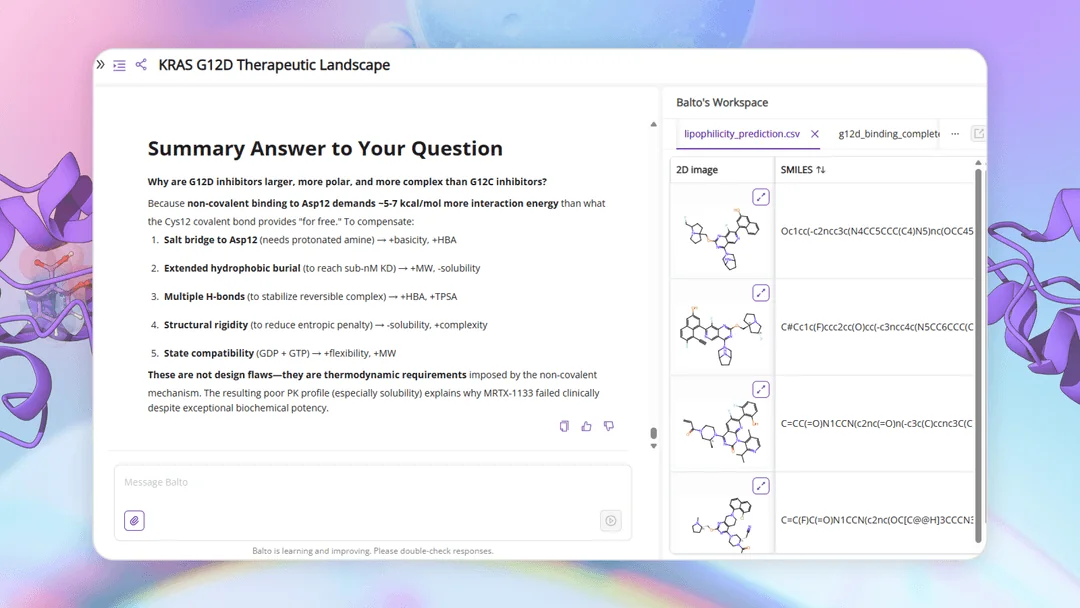
View Balto's full response
KRAS G12D as a Drug Target: Current Biological and Therapeutic Landscape
Overview
KRAS G12D represents the most prevalent KRAS mutation in pancreatic ductal adenocarcinoma (PDAC, ~40% of cases) and occurs frequently in colorectal cancer (CRC) and non-small cell lung cancer (NSCLC). After decades of being considered “undruggable,” KRAS G12D has emerged as a tractable target through multiple mechanistic approaches, though no agent has yet achieved regulatory approval as of January 2026.
Validated Mechanisms of Inhibition
1. Non-Covalent Allosteric Inhibition (RAS-OFF State)
MRTX1133 pioneered direct G12D targeting via high-affinity, non-covalent binding to the switch-II pocket of GDP-loaded KRAS G12D. This approach stabilizes the inactive conformation, blocking nucleotide exchange and effector binding with sub-nanomolar potency and strong allele selectivity (Cancer Discovery, PMID: 36216931).
Preclinical achievements: MRTX1133 demonstrated widespread tumor regressions in PDAC xenografts and immunocompetent models, reprogrammed the tumor microenvironment by increasing CD8+ T-cell infiltration and FAS expression, and synergized with PD-1/PD-L1 checkpoint blockade (Immunity, PMID: 37625401).
Clinical outcome: The Phase 1/2 trial (NCT05737706) was terminated in March 2025 after Phase 1 due to highly variable, suboptimal pharmacokinetics—not safety concerns—highlighting translational challenges for non-covalent switch-II pocket binders in humans.
2. Covalent RAS-ON Inhibition via Tri-Complex (Molecular Glue) Catalysis
Zoldonrasib (RMC-9805) represents a paradigm shift: it acts as a molecular glue, recruiting cyclophilin A to form a neomorphic protein-protein interface with GTP-bound (active) KRAS G12D. This induced proximity catalyzes selective, irreversible covalent modification of the Asp12 residue, disabling downstream signaling (Science, July 2025; company announcement).
Clinical progress: As of January 2026, zoldonrasib holds FDA Breakthrough Therapy Designation for previously treated KRAS G12D-mutant NSCLC, based on a 61% objective response rate and 89% disease control rate at the 1200 mg QD dose in early-phase trials (AACR 2025; FDA announcement January 8, 2026). This is the most clinically advanced G12D-targeted therapy to date.
3. Targeted Protein Degradation (PROTACs)
Multiple KRAS G12D-selective degraders have been reported in 2024-2025, representing an event-driven pharmacology approach that eliminates the mutant protein entirely:
- Compound 8o (J. Med. Chem. 2024): Built on MRTX1133 scaffolds, induced rapid, selective G12D degradation with strong pathway suppression and significant antitumor efficacy in AsPC-1 PDAC xenografts.
- CH091138 (Eur. J. Med. Chem. 2025): Selectively degrades endogenous KRAS G12D (not WT or other mutants), downregulates KRAS in proteomics, and suppresses AsPC-1 tumors and G12D patient-derived organoids in a VHL- and proteasome-dependent manner.
- ZJK-807 (J. Med. Chem. 2025): Overcomes resistance mutations affecting switch-II pocket inhibitors with selective cellular degradation and in vivo tumor growth inhibition.
4. Direct Covalent Targeting via Strain-Release Electrophiles
A 2024 chemistry breakthrough demonstrated that malolactone-based, strain-release electrophiles grafted onto switch-II pocket ligands can react selectively with the Asp12 carboxylate, forming stable covalent adducts in both GDP and GTP states (Nature Chemical Biology, 2024).
Key Signaling Pathways Involved
RAF→MEK→ERK (MAPK) Axis
KRAS G12D drives constitutive activation of the MAPK cascade, which is essential for PDAC initiation and maintenance. ERK drives the majority of the KRAS-dependent phosphoproteome in KRAS-mutant PDAC.
PI3K→AKT→mTORC1 Axis
KRAS G12D directly engages class I PI3Ks with enhanced affinity compared to wild-type KRAS, activating survival and metabolic reprogramming pathways.
Crosstalk and Adaptive Signaling
Loss of ERK-mediated negative feedback drives rebound signaling through wild-type RAS isoforms to both ERK and AKT. In KRAS G12D-mutant CRC, MRTX1133 triggers EGFR/pan-ERBB feedback that activates wild-type RAS and rescues MAPK and PI3K signaling.
Clinical and Preclinical Failures & Resistance Mechanisms
Clinical Failures to Date:
- MRTX1133: Despite exceptional preclinical potency, the Phase 1/2 trial was terminated due to highly variable, suboptimal human PK without a safety signal.
- Checkpoint blockade monotherapy: Limited benefit in PDAC outside MSI-H/dMMR subsets (~1-2% of cases).
Mechanisms of Resistance:
- MAPK pathway reactivation via RTKs/EGFR/ERBB
- Upstream RAS reactivation through SHP2/SOS1
- Bypass signaling and parallel pathway engagement (PI3K/AKT/mTOR)
- Metabolic and cell-state adaptations (autophagy-mediated glutathione synthesis)
- Pocket and interface mutations (Q95/Y96 conferring resistance to pocket-binding inhibitors)
- Tumor microenvironment and immune evasion
Open Questions and Emerging Strategies
Promising but Underexplored Approaches:
- KRAS G12D inhibition + immunotherapy combinations in PDAC
- Neoantigen vaccines at minimal residual disease (ELI-002)
- Combination with stromal/chemokine remodeling
- Optimizing tri-complex and molecular glue strategies
- Multi-epitope TCR-T and HLA-loss countermeasures
Before touching a molecule, discovery starts with context.
Is the target druggable?
Which mechanisms have worked — and which have failed?
Where does resistance emerge, and what strategies might overcome it?
Rather than issuing a single broad search, Balto approaches this step the way an experienced scientist would: by iteratively refining its questions as new information emerges.
The initial prompt asked Balto to summarize the therapeutic landscape around KRAS G12D — not just listing inhibitors, but identifying validated mechanisms, clinical outcomes, resistance pathways, and open questions. From there, Balto decomposed the task into a sequence of focused research passes.
In response, Balto began by surveying known KRAS G12D inhibitors, then pivoted to clinical trial outcomes, followed by mechanistic studies explaining both success and failure. As resistance patterns emerged, Balto expanded its search to upstream and downstream signaling pathways. Finally, it explored emerging strategies — molecular glues, degraders, immunotherapy combinations — that attempt to address the limitations of first-generation approaches.
Internally, this looked like a loop of refinement:
- initialize session memory
- search inhibitor literature
- assess clinical success and failure
- analyze resistance mechanisms
- map signaling pathways and adaptive feedback
- explore emerging and underexplored strategies
- update memory with consolidated findings
Each step informed the next. Rather than treating papers as isolated facts, Balto integrated them into a growing mental model of the target. It then saved these observations and supporting data to its memory to build on throughout the subsequent steps.
To put this another way, the result is not a flat literature summary, but a structured view of the KRAS G12D landscape: which mechanisms are validated, where pharmacology has broken down, how tumors adapt, and where new opportunities may lie.
To make this information usable beyond the chat, Balto also exports ranked results — including top pathway papers, inhibitor studies, and clinical outcomes — as CSVs for further inspection. Chemists can scan, sort, and cross-reference these outputs just as they would any internal dataset.
In practice, this step compresses weeks of background research into minutes — giving scientists a clear foundation to decide which mechanisms are worth pursuing, and setting up the next step: identifying and comparing known active molecules.
Step 2 — Finding known actives and mechanistic anchors
Our prompt to Balto:
Using ChEMBL, identify known small molecules that directly target KRAS G12D or modulate KRAS signaling via SOS1 or SHP2.
For each compound, return:
- Target and mechanism
- Mutation or allele specificity
- Best reported biochemical and cellular potencies
- Assay context
- Development stage
Rank results by relevance to KRAS G12D biology and strength of evidence.
View Balto's full response
Summary of what I delivered:
- 538 KRAS G12D-specific activities analyzed
- 100 SOS1-KRAS inhibitors identified
- 200 SHP2 bioactivity records compiled
- Ranked by relevance to KRAS G12D biology and evidence strength
- 12 top compounds profiled with all requested details
Key findings from the analysis:
Direct KRAS G12D Inhibitors:
- MRTX-1133: 0.4 nM IC50, Phase 1 (terminated due to PK issues)
- CHEMBL4855757: 0.1 nM IC50 (ultra-potent, preclinical)
SOS1 Inhibitors:
- BI-1701963: Phase 1 active, pan-KRAS mechanism
- MRTX0902: Phase 1, brain-penetrant, deprioritized by BMS
- Top ChEMBL compounds: 1–2 nM IC50
SHP2 Inhibitors:
- JAB-3312: Phase III (China), ORR 65–71% with glecirasib
- TNO155: Phase 1b/2, ORR ~33% with JDQ443
- Top ChEMBL compounds: 0.32–7 nM IC50
#1 by Pure Biochemical Potency: CHEMBL4855757
IC50: 0.1 nM (most potent G12D inhibitor in ChEMBL)
Limitations:
- Only binding data - no cellular functional assay
- No published name or structure disclosed
- No mechanism details beyond G12D binding
- No in vivo or clinical data
#1 by Potency + Mechanism Quality: MRTX-1133 (CHEMBL4858364) — RECOMMENDED
Biochemical IC50: 0.4 nM | Cellular IC50: 50 nM
Target & Mechanism: Non-covalent, allosteric inhibitor that binds the switch-II pocket of GDP-loaded KRAS G12D.
Why MRTX-1133 is the better #1:
- Functional validation - works in cells, not just in a test tube
- Defined mechanism - switch-II pocket allosteric inhibitor with structural basis
- Clinical-grade - advanced to human trials with extensive preclinical validation
- Comprehensive dataset - biochemical + cellular + in vivo + TME remodeling studies
Preclinical Achievements:
- Tumor regressions in PDAC xenografts and syngeneic models
- Reprograms tumor microenvironment (↑CD8+ T cells, ↑FAS, ↓myeloid)
- Synergizes with PD-1/PD-L1 checkpoint blockade
- Combination-ready: validated with anti-EGFR, SOS1i, autophagy inhibitors
With a biological hypothesis in hand, the next question is not “what molecules exist?” — it’s “which chemical strategies have already demonstrated real traction against this biology?”
Rather than issuing a narrow keyword search, the Step 2 prompt explicitly told Balto to build on what it had already learned. That single instruction mattered. It signaled that this was a continuation of the same discovery thread, not an isolated lookup.
As a result, Balto reused its session memory from Step 1 — including validated mechanisms, resistance pathways, and translational challenges — to scope a focused ChEMBL analysis. It did not re-derive the KRAS G12D landscape. It went straight to identifying compounds that meaningfully intersect with it.
Internally, this meant querying ChEMBL across multiple mechanistic classes in parallel: direct KRAS G12D binders, SOS1 disruptors that suppress upstream signaling, and SHP2 inhibitors that blunt adaptive feedback. Each class was evaluated not just by potency, but by biological relevance — allele specificity, assay context, and evidence strength.
Instead of returning a long tail of loosely related hits, Balto collapsed hundreds of bioactivity records into a ranked short list. Across 538 KRAS G12D–specific activity entries, 100 SOS1 inhibitors, and 200 SHP2 records, it identified a dozen compounds that anchor the current state of the field — spanning ultra-potent preclinical binders, clinically tested pathway modulators, and combination-validated SHP2 programs.
Crucially, Balto treated these compounds as mechanistic exemplars, not just names on a list. For each, it surfaced how the molecule works, where it has succeeded or failed clinically, and what limitations remain unresolved. In doing so, it turned ChEMBL from a database into a decision-making tool.
By the end of Step 2, the chemist isn’t just aware of what’s been made — they understand which chemical strategies have been validated, which have stalled due to pharmacology or resistance, and which modalities appear most promising for further exploration.
This sets a clear transition point. Once candidate molecules are identified and ranked by biological relevance, the next question becomes unavoidable:
What differentiates these compounds chemically — and which properties might explain their successes or failures?
Step 3 — Comparing physicochemical profiles and developability signals
Our prompt to Balto:
Building on the top KRAS G12D-relevant compounds identified in the previous step, retrieve physicochemical properties and bioactivity summaries from PubChem
For each compound include:
- Molecular weight, cLogP, TPSA, HBD/HBA, rotatable bonds
- Aqueous solubility and permeability indicators (if available)
- Reported bioavailability, formulation notes, and exposure-related flags
- SMILES strings for downstream modeling and docking
Benchmark these properties against sotorasib and adagrasib, and highlight which parameters may explain known pharmocokinetic limitations or clinical outcomes
View Balto's full response
Physicochemical & ADMET Profile Analysis: Top KRAS Compounds
Complete Property Comparison Table
| Property | CHEMBL4855757 | MRTX-1133 | Sotorasib | Adagrasib | Ideal Range |
|---|---|---|---|---|---|
| Clinical Stage | Preclinical | Phase 1 (terminated) | FDA Approved | FDA Approved | — |
| Molecular Weight | 572.3 Da | 600.2 Da ⚠️ | 560.6 Da ✓ | 604.1 Da ⚠️ | <500 Da |
| cLogP | 3.56 ✓ | 3.91 ⚠️ | 3.37 ✓ | 3.48 ✓ | 0-3 |
| LogS (solubility) | -4.74 ⚠️ | -4.78 ⚠️ | -3.99 ✓ | -4.06 ✓ | >-4 |
| HBA (acceptors) | — | 11 ⚠️ | 7 ✓ | 9 ⚠️ | ≤10 |
| QED Score | 0.33 ⚠️ | 0.32 ⚠️ | 0.36 ⚠️ | 0.36 ⚠️ | >0.5 |
Key Findings: What Explains PK Limitations?
MRTX-1133 PK Failure Root Causes
The clinical termination of MRTX-1133 due to “variable/suboptimal PK” can be explained by multiple compounding factors:
A. Molecular Weight & Complexity Burden
- MW = 600.2 Da (20% above Lipinski’s Rule of 500 Da)
- High polarity: 11 hydrogen bond acceptors (vs. 7 for sotorasib)
- Result: Poor oral absorption, high first-pass metabolism, variable bioavailability
B. Solubility Crisis
- LogS = -4.78 (predicted aqueous solubility ~10 μM)
- Sotorasib LogS = -3.99 (65 μM, 6.5× more soluble)
C. Poor Drug-Likeness
- QED = 0.32 (where 1.0 = ideal drug)
- Lower than sotorasib (0.36) and adagrasib (0.36)
Why Sotorasib and Adagrasib Succeeded
Despite also being large, complex molecules, the approved G12C inhibitors have critical advantages:
Sotorasib (Lumakras) - Superior Profile:
- Better solubility: LogS -3.99 (6.5× > MRTX-1133)
- Lower MW: 560.6 Da (40 Da lighter)
- Fewer HBA: 7 vs. 11 (better permeability)
- Optimal cLogP: 3.37 (better absorption/distribution balance)
Adagrasib (Krazati) - Acceptable Compromises:
- Similar solubility: LogS -4.06 (slightly better than MRTX-1133)
- No HBD: 0 donors (reduces crystal lattice energy, aids dissolution)
- Required advanced formulation strategy (amorphous solid dispersion)
- Higher dose to compensate (600 mg BID vs QD)
Recommendations for G12D Inhibitor Optimization
Priority 1: Improve Solubility — Target LogS > -4.0
Priority 2: Reduce Molecular Weight — Target MW < 550 Da
Priority 3: Optimize HBA Count — Target ≤8 HBA
Priority 4: Maintain Potency — Accept modest potency loss (5-10×) for 10× better PK
By this point, Balto has already identified which chemical strategies matter biologically.
Step 3 asks a harder — and more consequential — question:
Why do some of these molecules translate clinically, while others stall despite extraordinary potency?
Rather than treating PubChem as a static property lookup, the Step 3 prompt explicitly instructed Balto to build on the ranked compounds from Step 2 and benchmark them against known clinical outcomes. That framing mattered. It told Balto that this step was about explanation, not enumeration.
As a result, Balto didn’t simply return molecular weights and LogP values. It assembled a comparative, decision-oriented analysis — pulling physicochemical properties, estimating ADMET risk, and contextualizing each value against real-world outcomes for approved and failed programs.
Internally, Balto treated sotorasib and adagrasib as calibration points, not the main event. Their property profiles were used to anchor expectations: what ranges proved workable in humans, what compromises were tolerated, and which formulation strategies ultimately succeeded. Against that backdrop, Balto evaluated next-generation KRAS G12D inhibitors — including MRTX-1133 and ultra-potent preclinical binders — and highlighted where physicochemical red flags accumulated.
This is where the workflow moves beyond “potency-first thinking.”
Balto surfaced how multiple small disadvantages compound:
- marginal solubility
- excess molecular weight
- high hydrogen bond acceptor counts
- structural complexity
Together, these explain why a molecule like MRTX-1133 could demonstrate breathtaking preclinical efficacy — yet fail in the clinic due to variable and suboptimal exposure.
Crucially, Balto didn’t stop at diagnosis. It translated these observations into actionable medicinal chemistry guidance: which parameters are most predictive of failure, which tradeoffs are hardest to fix, and where modest potency loss might be justified to achieve viable pharmacokinetics.
By the end of Step 3, the chemist has more than a property table. They have a coherent hypothesis linking mechanism, structure, physicochemical profile, and clinical outcome — along with a clear sense of which candidates are worth pushing forward, and which require rethinking at the scaffold level.
At this point, one question naturally remains:
If these molecules are so large and complex, what exactly are they doing in the binding pocket — and where is that complexity coming from?
Answering that requires looking directly at experimental binding evidence and structure.
Step 4 — Explaining binding-driven complexity
Our prompt to Balto:
Using available experimental data, compare how leading KRAS inhibitors bind the switch-II pocket.
Specifically:
- Show biochemical binding data for sotorasib, adagrasib, MRTX-1133, and representative KRAS G12D inhibitors
- Retrieve associated PDB structures or cryo-EM models
- Highlight key ligand–protein interactions that drive affinity, selectivity, and residence time
- Explain how differences in G12C vs G12D binding mechanisms impose distinct structural and physicochemical constraints
Focus on how binding mode explains the molecular size, polarity, and complexity observed in G12D inhibitors.
View Balto's full response
Comprehensive Structural Analysis: KRAS Inhibitor Binding Modes
Biochemical Binding Data Summary
| Parameter | Sotorasib (G12C) | Adagrasib (G12C) | MRTX-1133 (G12D) |
|---|---|---|---|
| Mechanism | Covalent GDP-state | Covalent GDP-state | Non-covalent (GDP/GTP) |
| KD (pre-covalent) | ~220 nM | 9.59 ± 2.09 nM | 0.40 ± 0.11 nM |
| Residence Time | Protein turnover | Protein turnover | Est. 4-40 min |
| Selectivity (WT) | ~700-fold | >6,000-fold | ~6,000-fold |
Key Insight: G12C covalent inhibitors benefit from permanent engagement (residence time = protein lifetime), while G12D non-covalent inhibitors must achieve sub-nanomolar KD with reversible binding.
Crystal Structure Analysis
| Compound | PDB ID | Resolution | Key Features |
|---|---|---|---|
| Sotorasib | 6OIM | 1.65 Å | Covalent Cys12; H95/Y96/Q99 cryptic pocket |
| Adagrasib | 6UT0 | 1.94 Å | Covalent Cys12; His95 H-bond; 8-Cl-naphthyl deep pocket |
| MRTX-1133 | 7T47 | 1.27 Å | Non-covalent; Asp12 salt bridge; state-compatible |
Why G12D Inhibitors Are Larger: The Non-Covalent Affinity Problem
Covalent G12C Inhibitors:
- Pre-covalent KD can be 10-200 nM (moderate)
- Covalent bond adds ~5-7 kcal/mol “free” binding energy
- Post-covalent residence time = protein lifetime (hours to days)
- Result: Can use smaller, less complex scaffolds
Non-Covalent G12D Inhibitors:
- Must achieve KD < 1 nM WITHOUT covalent bond
- Requires ~10 kcal/mol total binding energy from:
- Salt bridge (Asp12): ~4-5 kcal/mol
- Hydrophobic burial: ~3-4 kcal/mol
- Multiple H-bonds: ~2-3 kcal/mol
- Shape complementarity: ~1-2 kcal/mol
- Complex dissociates on minutes timescale
- Result: Requires larger scaffold with more interaction points
The G12D Druggability Paradox
Non-Covalent Mechanism REQUIRES: → Extensive Interaction Network (to achieve <1 nM KD) → Large Molecular Size (>550 Da) + High Polarity (>10 HBA) → Poor Solubility + Poor Permeability → Challenging PK Profile → Clinical Translation Risk
Not a chemistry failure—an inevitable consequence of non-covalent G12D targeting.
Path Forward for G12D Inhibitors
Three Viable Strategies:
-
Tri-Complex Covalent (Zoldonrasib): Reintroduces covalency via CypA catalysis. Early clinical success (FDA BTD).
-
Targeted Protein Degradation (PROTACs): Circumvents pocket mutations. Event-driven pharmacology.
-
Formulation Innovation: Advanced formulations for existing scaffolds. Higher risk path.
By the end of Step 3, the picture was clear — and unsettling.
The most potent KRAS G12D inhibitors were not failing because of sloppy chemistry or avoidable mistakes. Their physicochemical liabilities were structurally imposed. The question was no longer whether these molecules were “too big” or “too polar,” but why they had to be.
Step 4 turns to binding evidence to answer that question directly.
Using BindingSearch, Balto aggregated experimental binding data alongside available structural models for leading KRAS inhibitors — including approved G12C drugs (sotorasib and adagrasib), clinical-stage G12D inhibitors (MRTX-1133), and representative high-potency G12D binders from the literature. This allowed potency, residence time, and binding mode to be examined together, rather than in isolation.
What emerged was a clear mechanistic explanation for everything uncovered earlier.
Covalent G12C inhibitors benefit from a privileged anchor: Cys12. That single electrophilic bond compensates for lower intrinsic affinity, allowing smaller, more drug-like molecules to achieve durable inhibition with fewer interactions and reduced structural burden. In contrast, G12D inhibitors lack a comparable covalent handle. To achieve slow off-rates and sufficient pathway suppression, they must rely on extensive non-covalent interactions across the switch-II pocket.
Those interactions come at a cost.
Balto highlighted how G12D binders require larger contact surfaces, more hydrogen bond acceptors, and tighter geometric complementarity — all of which translate directly into increased molecular weight, polarity, and rigidity. The very features that enable sub-nanomolar binding also degrade solubility, permeability, and oral exposure.
In other words, the physicochemical problems observed in Step 3 were not accidental. They were the downstream consequence of binding mode.
This structural perspective closes the loop on the investigation. It explains why certain optimization strategies struggle, why formulation alone often isn’t enough, and why alternative modalities — such as tri-complex “molecular glue” approaches or event-driven degradation — are gaining traction for KRAS G12D.
By the end of Step 4, the chemist isn’t just comparing compounds. They understand the mechanistic tradeoffs imposed by the target itself — and can reason about which strategies are likely to scale, and which will continue to fight the physics of the pocket.
Bringing it together: from hypothesis to a defensible lead
Drug discovery rarely fails because scientists lack ideas.
It fails because connecting those ideas to evidence takes too long to vet and push the truly good ideas forward.
In this walkthrough, we didn’t try to design a drug. We did something more fundamental: we compressed early discovery thinking — the part that usually lives across dozens of tabs, spreadsheets, and half-finished notes — into a single, coherent flow.
Step by step, Balto acted less like a search engine and more like a junior scientist who remembers what they’ve learned.
- With LiteratureSearch and WebResearch, Balto mapped the biological landscape: mechanisms, pathways, resistance, and open questions — not just what’s known, but what still matters.
- With CHEMBLSearcher, it grounded that biology in chemistry, identifying which molecular strategies have actually shown traction and which have failed for reasons beyond potency.
- With PubChemSearch, Balto explained why those failures occurred, linking physicochemical properties to real-world pharmacokinetic outcomes rather than treating properties as abstract numbers.
- And with BindingSearch, it closed the loop structurally — showing how binding mode drives molecular complexity, selectivity, and ultimately developability.
By the end of the process, we didn’t just have a list of compounds (though we also gained this). We had something far more valuable:
- a clear biological rationale
- a ranked set of mechanistically relevant molecules
- an understanding of why certain approaches succeed or stall
- and a defensible hypothesis for what a promising next molecule would need to look like — chemically and biologically
That’s the real outcome of early discovery.
Balto doesn’t replace medicinal chemistry judgment. It accelerates the part of the work where judgment matters most — by ensuring that every decision is grounded in integrated evidence rather than fragmented searches.
This first tutorial focused on finding promising molecules and their biological background. In future walkthroughs, we’ll build directly on this foundation — moving into structure-guided optimization, simulation, docking, and ADMET prediction — without ever having to restart from scratch.
Because discovery doesn’t happen in steps. It compounds.