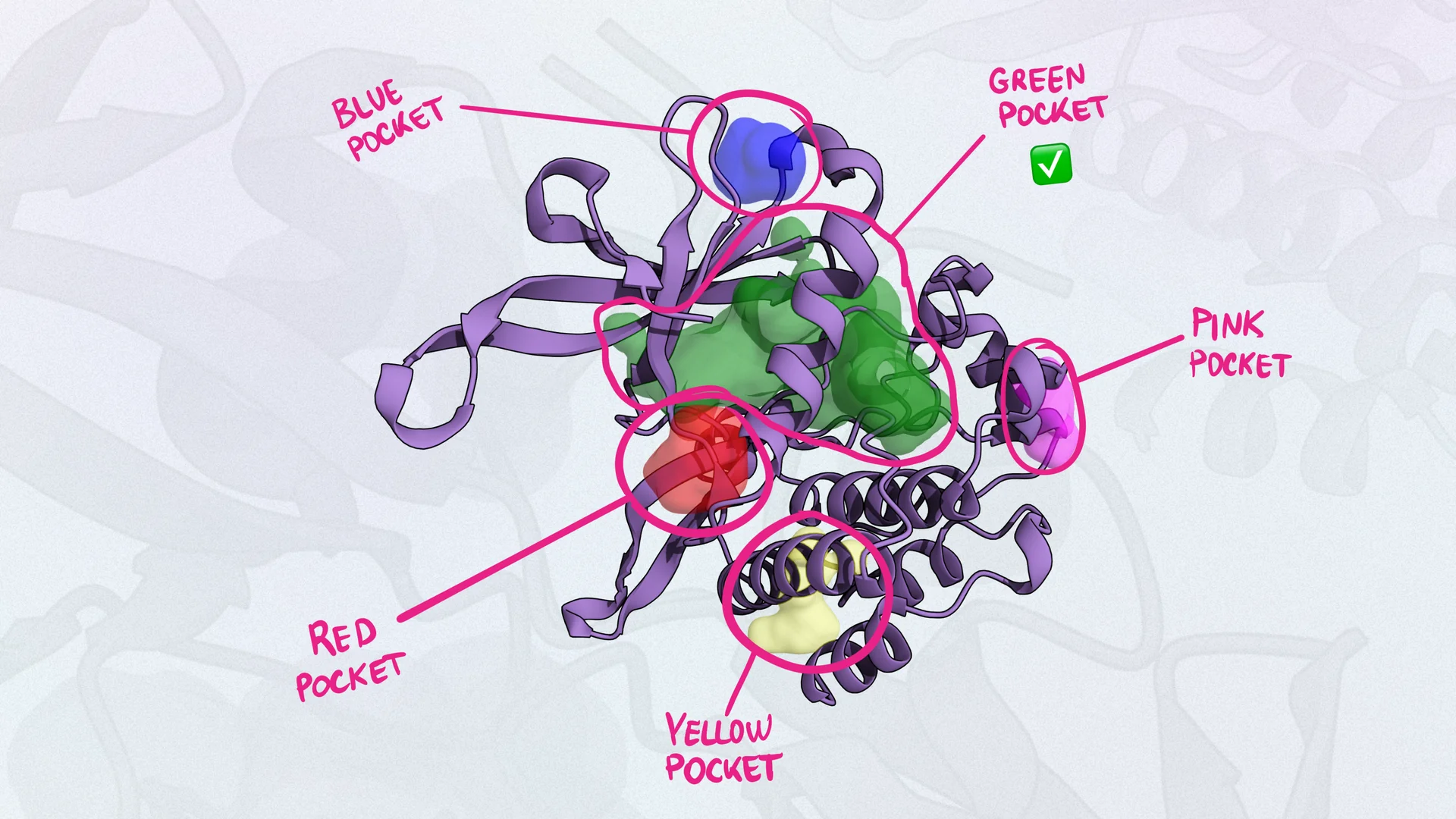
Pharmacophore Modeling
methodPharmacophore modeling is used to identify and represent the essential features of a molecule that are necessary for binding or activity. These features, known as pharmacophores, include hydrogen bond acceptors and donors, hydrophobic regions, aromatic rings, and charged groups. A pharmacophore model is a three-dimensional arrangement of these features that can be used to filter molecules to interact with a specific biological target to produce a desired effect.
Importance in Computational Drug Discovery
- Target Identification: Pharmacophore models help identify the key interaction points between a ligand and its biological target, facilitating the understanding of the molecular basis of ligand binding.
- Virtual Screening: Pharmacophore models can be used to screen large libraries of compounds to identify potential drug candidates that possess the required pharmacophoric features.
- Lead Optimization: By highlighting essential features for activity, pharmacophore models aid in the optimization of lead compounds to enhance their potency and selectivity.
Key Tools
-
Pharmit: An online platform for pharmacophore modeling and virtual screening.
-
LigandScout: A software tool for creating and applying pharmacophore models.
-
MOE (Molecular Operating Environment): A comprehensive suite for molecular modeling, including pharmacophore modeling and virtual screening.
-
Discovery Studio: A software platform offering tools for pharmacophore modeling, docking, and molecular dynamics simulations.
Literature
“Pharmacophore Modeling in Drug Discovery: Methodology and Current Status”
-
Publication Date: 2021-06-29
-
Summary: Reviews the methodology of pharmacophore modeling, its integration with other computational methods, and its applications in drug discovery.
“Pharmacophore Modeling in Drug Discovery and Development: An Overview”
-
Publication Date: 2007-02-28
-
Summary: Provides a historical overview of pharmacophore modeling and discusses developments in methodologies for pharmacophore identification and their applications in drug discovery.
“A Computer-Aided Drug Discovery Based Discovery of Lead-Like Compounds Against KDM5A for Cancers Using Pharmacophore Modeling and High-Throughput Virtual Screening”
-
Publication Date: 2021-10-12
-
Summary: Identifies lead compounds for KDM5A through pharmacophore modeling and high-throughput virtual screening, with further evaluation using ADMET properties and molecular dynamics simulations.
“The Development of Pharmacophore Modeling: Generation and Recent Applications in Drug Discovery”
-
Publication Date: 2018-12-08
-
Summary: Reviews successful examples of pharmacophore modeling applied in virtual screening and lead optimization, providing an overview of pharmacophore-based virtual screening.
“Pharmacophore Modeling: Advances, Limitations, and Current Utility in Drug Discovery”
-
Publication Date: 2014-11-11
-
Summary: Reviews the computational implementation of the pharmacophore concept and its common usage in drug discovery, including virtual screening, ADME-tox modeling, and target identification.
“Computational Discovery of SARS-CoV-2 NSP 16 Drug Candidates Based on Pharmacophore Modeling and Molecular Dynamics Simulation”
-
Publication Date: 2021-06-01
-
Summary: Uses pharmacophore-based virtual screening and molecular dynamics simulations to identify potential SARS-CoV-2 NSP 16 inhibitors.
“Azolium Analogues as CDK4 Inhibitors: Pharmacophore Modeling, 3D QSAR Study and New Lead Drug Discovery”
-
Publication Date: 2017-04-15
-
Summary: Presents ligand-based pharmacophore modeling and 3D-QSAR analyses for azolium-based CDK4 inhibitors.
“The Discovery of Novel BCR-ABL Tyrosine Kinase Inhibitors Using a Pharmacophore Modeling and Virtual Screening Approach”
-
Publication Date: 2021-03-04
-
Summary: Identifies novel BCR-ABL inhibitors through pharmacophore modeling and virtual screening, with in vitro validation.
“Unlocking Neuraminidase Inhibitors: Insights from Natural Products through Pharmacophore Modeling, Virtual Screening, and Molecular Docking”
-
Publication Date: 2024-11-08
-
Summary: Identifies potential neuraminidase inhibitors from natural products using pharmacophore modeling, virtual screening, and molecular docking.

 to discover more
to discover more Explore more materials, our research and analyses of AI and the drug discovery industry below.